อ.พญ. ชมชนัท ทับเจริญ
รศ.พญ. วรนุช จงศรีสวัสดิ์
สาขาวิชาโรคทางเดินอาหารและตับ
ภาควิชากุมารเวชศาสตร์ คณะแพทยศาสตร์ จุฬาลงกรณ์มหาวิทยาลัย
โรคตับเรื้อรังในผู้ใหญ่เป็นที่รู้จักแพร่หลาย ซึ่งสาเหตุส่วนใหญ่มักเกิดจากการดื่มแอลกอฮอล์ การติดเชื้อไวรัสตับอักเสบบีหรือซี ส่วนโรคตับในเด็กมักไม่เป็นที่รู้จักในประชาชนส่วนใหญ่รวมทั้งอายุรแพทย์ สาเหตุของโรคตับเรื้อรังในเด็กแตกต่างกับในผู้ใหญ่และมีความหลากหลายในแต่ละช่วงอายุ กล่าวคือ ช่วงทารกและเด็กเล็กสาเหตุมักเกิดจากความผิดปกติแต่กำเนิดหรือความผิดปกติทางพันธุกรรม ตัวอย่างเช่น ความผิดปกติของท่อน้ำดีภายในหรือภายนอกตับ ได้แก่ โรคท่อน้ำดีตีบตัน (biliary atresia) โรคท่อน้ำดีโป่งพอง (choledochal cyst) และ Alagille syndrome ความผิดปกติของการขับน้ำดี ได้แก่ progressive familial intrahepatic cholestasis (PFIC) ความผิดปกติของการสร้างน้ำดี ได้แก่ bile acid synthesis defects โรคตับเมแทบอลิกหรือโรคในกลุ่มเมแทบอลิซึมผิดปกติแต่กำเนิด (inborn error of metabolism) เป็นต้น ส่วนโรคตับเรื้อรังในเด็กโตและวัยรุ่นอาจเกิดจากการติดเชื้อไวรัสตับอักเสบบีหรือซี การได้รับยาหรือสารพิษ โรคภูมิต้านทานต่อตับตนเอง (autoimmune liver disease) และตับคั่งไขมันที่ไม่ได้เกิดจากการดื่มแอลกอฮอล์ เป็นต้น ภาวะแทรกซ้อนของโรคตับเรื้อรังในเด็กคล้ายกับในผู้ใหญ่ได้แก่ ascites, hypersplenism, malnutrition, edema และ variceal bleeding1,2
ภาวะตัวเหลืองในทารกอายุมากกว่า 2 สัปดาห์เป็นภาวะที่พบได้บ่อย ส่วนใหญ่เกิดจาก breast milk jaundice ซึ่งไม่รุนแรงและหายเองได้ แต่มีทารกประมาณ 1 ต่อ 2500 ของทารกเกิดมีชีพที่เป็นตัวเหลืองจากภาวะน้ำดีคั่ง (cholestasis) โดยสาเหตุที่พบบ่อยที่สุดในกลุ่มนี้ คือ โรคท่อน้ำดีตีบตัน3 ในบทความนี้จะกล่าวถึงเฉพาะโรคท่อน้ำดีตีบตันและ Alagille syndrome ซึ่งอาจมีความสับสนในการวินิจฉัยแยก 2 โรคนี้ได้
โรคท่อน้ำดีตีบตัน (biliary atresia)
เป็นโรคที่มีการอักเสบและเกิดพังผืดที่ท่อน้ำดีจึงทำให้มีการตีบตันอย่างถาวร ทำให้น้ำดีที่สร้างจากตับไม่สามารถไหลลงสู่ลำไส้เล็กได้ ในเอเชียพบอุบัติการณ์ประมาณ 1 ต่อ 5,000 ถึง 10,000 ของทารกเกิดมีชีพ และพบในเพศหญิงมากกว่าเพศชายเล็กน้อย4 ปัจจุบันยังไม่ทราบสาเหตุที่แน่ชัดของการเกิดโรค สันนิษฐานว่าอาจเกิดจากปัจจัยดังต่อไปนี้ ได้แก่ ความผิดปกติแต่กำเนิดของการสร้างทางเดินน้ำดี (congenital malformation), การติดเชื้อไวรัสบางชนิด เช่น reovirus, rotavirus, Epstein-Barr virus หรือ cytomegalovirus, ความผิดปกติของระบบภูมิคุ้มกันหรือพันธุกรรม เป็นต้น โรคนี้แบ่งตามตำแหน่งของท่อน้ำดีที่ตีบตันเป็น 3 ชนิด ดังแสดงในรูปที่ 1
รูปที่ 1. ชนิดของโรคท่อน้ำดีตีบตัน โดยสีดำหมายถึงทางเดินน้ำดีที่ตีบตัน ส่วนสีเทาหมายถึงทางเดินน้ำดีที่น้ำดีไหลผ่านได้ และสีขาวหมายถึงทางเดินน้ำดีที่ไม่มีน้ำดีไหลผ่าน (ได้รับความอนุเคราะห์รูปจากศาสตราจารย์ ดร. นายแพทย์ไพศาล เวชชพิพัฒน์)
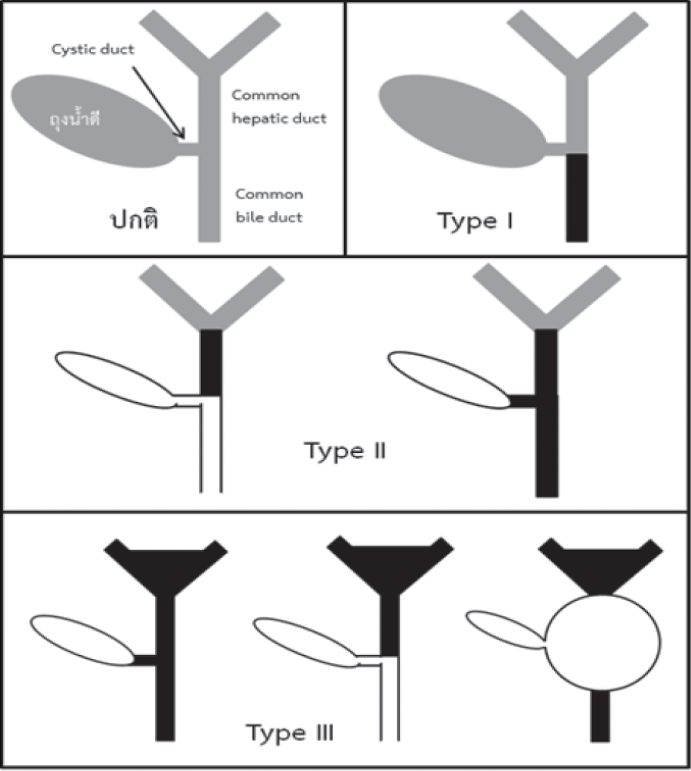
อาการแสดงที่พบ คือ ทารกจะมีอาการตัวหรือตาเหลืองช่วงอายุ 2-3 สัปดาห์แรกร่วมกับอุจจาระสีซีดหรือเหลืองอ่อน ปัสสาวะสีเหลืองเข้ม ในรายที่โรคดำเนินต่อโดยไม่ได้รับการรักษาที่เหมาะสมจะพบภาวะตับแข็ง บางรายมีไข้ต่ำๆ ท้องโตจากน้ำในท้องหรือม้ามโต หรือมีอุจจาระเป็นมัน (steatorrhea)
การตรวจค่าการทำงานของตับ พบระดับ total/direct bilirubin และ gamma-glutamyltransferase (GGT) สูง อย่างไรก็ตามผลการตรวจดังกล่าวไม่เพียงพอต่อการแยกโรคท่อน้ำดีตีบตันออกจากโรคอื่นที่เป็นสาเหตุให้เกิดภาวะน้ำดีคั่งได้ จึงต้องส่งตรวจเพิ่มเติมทางรังสีวิทยาโดยการอัลตราซาวน์ช่องท้องด้านบน ลักษณะที่เข้าได้กับโรคท่อน้ำดีตีบตันมีดังนี้5 คือ พบเนื้อเยื่อผังพืดลักษณะรูปสามเหลี่ยม (triangular cord) บริเวณขั้วตับ (porta hepatis) ผนังหรือรูปร่างของถุงน้ำดีผิดปกติ ไม่เห็นท่อน้ำดีร่วม และมีเส้นเลือดแดงเฮพาติคขนาดใหญ่ อย่างไรก็ตามการตรวจวิธีนี้ต้องอาศัยความชำนาญของรังสีแพทย์ และในบางครั้งอาจไม่พบลักษณะดังกล่าวได้ชัดเจน จึงควรส่งตรวจเพิ่มเติมโดยการทำ hepatobiliary scintigraphy หรือ DISIDA scan เพื่อดูการขับน้ำดีลงสู่ลำไส้เล็กส่วนโดยใช้ไอโซโทป หากพบสารไอโซโทปผ่านลงมาที่ลำไส้สามารถยืนยันได้ว่าทารกไม่ได้เป็นโรคท่อน้ำดีตีบตัน แต่หากไม่พบจะไม่สามารถแยกโรคอื่นได้ เช่น severe neonatal hepatitis ที่ทำให้เซลล์ตับไม่สามารถขับสารไอโซโทปลงมาได้ ดังนั้นเมื่อการตรวจแบบไม่รุกล้ำข้างต้นไม่สามารถยืนยันการวินิจฉัยได้จึงมีความจำเป็นที่จะต้องใช้การตรวจที่รุกล้ำ ได้แก่ การตรวจชิ้นเนื้อตับ (percutaneous liver biopsy) และ/หรือ intra-operative cholangiography (IOC) เพื่อยืนยันการวินิจฉัย ลักษณะทางจุลพยาธิวิทยาของโรคท่อน้ำดีตีบตันประกอบด้วย portal tract fibrosis, ductular proliferation และ cholestasis with bile plugs
การรักษาโรคท่อน้ำดีตีบตันทำโดยการผ่าตัดระบายน้ำดีลงสู่ลำไส้ หรือ hepatic portoenterostomy (Kasai operation) ซึ่งควรทำภายในอายุ 60 วัน เพราะหากนานกว่านี้มีความเสี่ยงต่อการเกิดภาวะตับแข็ง หลังการผ่าตัดควรติดตามอาการผู้ป่วยร่วมกับตรวจดูค่าการทำงานของตับ พิจารณาการให้ยาต่าง ๆ เช่น วิตามินเอ ดี อี และเค ยาปฏิชีวนะ นอกจากนี้บางสถาบันยังมีการให้สเตียรอยด์ขนาดสูงหลังผ่าตัดเพื่อลดการอักเสบและการเกิดผังพืดที่รอยต่อของ portoenterostomy ซึ่งผลการศึกษาประสิทธิภาพของสเตียรอยด์ในการ clear jaundice หลังผ่าตัดยังไม่ชัดเจน6 ภาวะแทรกซ้อนที่พบบ่อยหลังผ่าตัด คือ การติดเชื้อในท่อน้ำดี (ascending cholangitis) ผู้ป่วยจะมีอาการไข้ ตัวเหลืองขึ้น อุจจาระซีดลง ซึ่งควรให้ยาปฏิชีวนะอย่างเหมาะสม เพราะหากเกิด ascending cholangitis ซ้ำ ๆ อาจทำให้ผลการผ่าตัดล้มเหลวและนำไปสู่ภาวะตับแข็งในที่สุด
การพยากรณ์โรคหลังการผ่าตัดแบ่งเป็น 3 กลุ่ม ได้แก่ กลุ่มที่หนึ่งเป็นกลุ่มที่ได้ผลดีหลังผ่าตัด พบประมาณร้อยละ 40-60 ผู้ป่วยมักหายเหลืองในช่วง 1-4 เดือนหลังผ่าตัด อย่างไรก็ตามระยะยาวก็อาจมีอาการของโรคตับเรื้อรังตามมา ผู้ป่วยที่ Professor Morio Kasai ผ่าตัดรักษาและมีอายุยืนยาวมากที่สุด ด้วย native liver คือ มีอายุมากกว่า 60 ปี7 กลุ่มที่สองเป็นกลุ่มที่ได้ผลปานกลาง พบประมาณร้อยละ 20-30 เป็นกลุ่มที่สามารถระบายน้ำดีได้หลังผ่าตัด แต่ตับบางส่วนเสื่อมสภาพไปแล้ว กลุ่มนี้จะยังมีตัวเหลืองอยู่ และค่อยๆ มีอาการของโรคตับเรื้อรังร่วมกับ portal hypertension ผู้ป่วยอาจต้องได้รับการผ่าตัดปลูกถ่ายตับในช่วงอายุ 2-18 ปี8 กลุ่มที่สามเป็นกลุ่มที่ผลการรักษาไม่ดี ตับเสียการทำงานอย่างมากและไม่สามารถระบายน้ำดีได้หลังผ่าตัด ผู้ป่วยมักเสียชีวิตหากไม่ได้รับการปลูกถ่ายตับในวัยทารก ปัจจุบันการผ่าตัดปลูกถ่ายตับในเด็กได้ผลเป็นที่น่าพอใจ จากข้อมูลของสมาคมปลูกถ่ายอวัยวะแห่งประเทศไทยในปี พ.ศ. 2559-2562 พบอัตราการรอดชีวิตที่ 1 ปีหลังการปลูกถ่ายตับร้อยละ 89 โดยโรคท่อน้ำดีตีบตันเป็นข้อบ่งชี้ที่พบบ่อยที่สุดในเด็ก (ร้อยละ 77.4)
Alagille syndrome9,10
เป็นโรคที่มีการถ่ายทอดทางพันธุกรรมแบบ autosomal dominant พบประมาณ 1 ใน 30,000 ของทารกเกิดมีชีพ เกิดจากความผิดปกติของ Notch signaling pathway โดยร้อยละ 95 มีการกลายพันธุ์ของยีน JAG1 และร้อยละ 2-4 มีการกลายพันธุ์ของยีน NOTCH2 ความสำคัญของโรคนี้ คือ ในวัยทารกผู้ป่วยอาจมีอาการแสดงและผลการตรวจชิ้นเนื้อตับพบ ductular proliferation คล้ายกับโรคท่อน้ำดีตีบตัน ประกอบกับยังมีลักษณะใบหน้าที่เป็น pathognomonic ไม่ชัดเจน ทำให้การวินิจฉัยอาจคลาดเคลื่อนได้ ผู้ป่วยบางรายได้รับการผ่าตัด Kasai operation โดยไม่จำเป็น ส่งผลให้มีความเสี่ยงต่อการเกิด ascending cholangitis และ progressive liver dysfunction
โรคนี้มีความรุนแรงของอาการทางตับหลากหลายร่วมกับมีอาการระบบอื่นด้วย ได้แก่ มีอาการคันอย่างมากโดยไม่สัมพันธ์กับระดับบิลิรูบินและจะเด่นชัดในช่วงอายุประมาณ 6 เดือน มีลักษณะหน้าตาจำเพาะ คือ หน้าผากโหนก จมูกแฟบ คางแหลมยื่น ใบหน้าทรงคล้ายสามเหลี่ยม เบ้าตาลึกและห่าง หัวใจมักพบผิดปกติแบบ peripheral pulmonary artery stenosis กระดูกสันหลังมีลักษณะ butterfly vertebra หรืออาจมีภาวะกระดูกบาง (osteopenia) ร่วมด้วย ตรวจตามักพบ posterior embryotoxon ซึ่งไม่มีผลกับการมองเห็น บางรายมีเลือดออกในสมองหรือ stroke จากหลอดเลือดผิดปกติ เช่น Moyamoya ผิวหนังมีการสะสมของก้อนไขมัน (xanthomas) โดยพบส่วนมากบริเวณมือและเข่า ซึ่งสัมพันธ์กับภาวะคอเลสเตอรอลในเลือดสูง บางรายพบการทำงานของท่อไตผิดปกติ เช่น tubular acidosis, renal dysplasia หรือมีพัฒนาการช้า เกณฑ์การวินิจฉัยใช้ลักษณะอาการแสดงที่พบดังกล่าวร่วมกับผลตรวจทางพยาธิวิทยาพบท่อน้ำดีในตับมีจำนวนน้อยลง (paucity of bile duct) ซึ่งหากตรวจก่อนอายุ 6 เดือนจะพบได้ร้อยละ 60 แต่หลังจากนั้นจะพบได้ร้อยละ 9511 หรือตรวจพบการกลายพันธุ์ของยีน JAG1 หรือ NOTCH2
ปัจจุบันยังไม่มีการรักษาที่จำเพาะ เป็นเพียงการรักษาประคับประคองภาวะน้ำดีคั่ง อาการคัน และให้โภชนาการที่เหมาะสม การดำเนินโรคและพยากรณ์ของโรคมีความหลากหลายขึ้นอยู่กับอวัยวะที่เกี่ยวข้อง ผู้ป่วยประมาณร้อยละ 75 ต้องได้รับการปลูกถ่ายตับก่อนอายุ 18 ปี12 โดยมีข้อบ่งชี้ คือ มีตับแข็งร่วมกับมีภาวะแทรกซ้อน หรือมีอาการคันที่ไม่สามารถรักษาประคับประคองได้ด้วยยา การศึกษาในต่างประเทศพบว่า อัตราการรอดชีวิตหลังปลูกถ่ายตับที่ 1 และ 5 ปี คือ ร้อยละ 82 และ 78 ตามลำดับ โดยพบการเสียชีวิตมากใน 30 วันแรกหลังปลูกถ่ายตับมากกว่ากลุ่มโรคท่อน้ำดีตีบตัน สาเหตุการตายเกิดจากภาวะแทรกซ้อนทางระบบประสาท ระบบหัวใจและหลอดเลือด และ graft failure13,14
สรุป
หากพบทารกที่ยังคงมีอาการตัวเหลืองหลังอายุ 2 สัปดาห์เป็นต้นไป ควรตรวจประเมินโดยละเอียด ในปัจจุบันมีความก้าวหน้าในการตรวจ next-generation DNA sequencing ช่วยทำให้สามารถวินิจฉัย intrahepatic cholestasis ในกลุ่ม hereditary cholestasis เช่น PFIC ได้ถูกต้องแม่นยำมากยิ่งขึ้น เพื่อนำไปสู่การดูแลรักษาที่เหมาะสม ผู้ป่วยเด็กโรคตับเรื้อรังมีอายุยืนยาวขึ้นจากการรักษาที่ดีขึ้นและการปลูกถ่ายตับ อายุรแพทย์จึงมีโอกาสที่จะได้ดูแลรักษาต่อไปเมื่อผู้ป่วยเด็กเหล่านี้เติบโตเป็นผู้ใหญ่
เอกสารอ้างอิง
- วรนุช จงศรีสวัสดิ์. โรคตับในเด็ก. กรุงเทพฯ: โรงพิมพ์แห่งจุฬาลงกรณ์มหาวิทยาลัย; 2561.
- Kriegermeier A, Green R. Pediatric cholestatic liver disease: Review of bile acid metabolism and discussion of current and emerging therapies. Front Med (Lausanne) 2020 May 5;7:149.
- Götze T, Blessing H, Grillhösl C, Gerner P, Hoerning A. Neonatal cholestasis – Differential diagnoses, current diagnostic procedures, and treatment. Front Pediatr 2015 Jun 17;3:43.
- Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA 2020 Mar 17;6:FSO466.
- Humphrey TM, Stringer MD. Biliary atresia: US diagnosis. Radiology 2007;244:845-51.
- Bezerra JA, Spino C, Magee JC, Shneider BL, Rosenthal P, Wang KS, et al. Use of corticosteroids after hepatoportoenterostomy for bile drainage in infants with biliary atresia: The START randomized clinical trial. JAMA 2014;311:1750-9.
- Kelay A, Davenport M. Long-term outlook in biliary atresia. Semin Pediatr Surg 2017;26:295-300.
- Bezerra JA, Wells RG, Mack CL, Karpen SJ, Hoofnagle JH, Doo E, et al. Biliary atresia: Clinical and research challenges for the Twenty-First century. Hepatology 2018;68:1163-73.
- Ayoub MD, Kamath BM. Alagille syndrome: Diagnostic challenges and advances in management. Diagnostics (Basel) 2020 Nov 6;10:907.
- Bass LM, Kamath BM. Inherited disorders of cholestasis in adulthood. Clin Liver Dis (Hoboken) 2013;2:200-3.
- Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis. Hepatology 1999;29:822-9.
- Kamath BM, Ye W, Goodrich NP, Loomes KM, Romero R, Heubi JE, et al. Outcomes of childhood cholestasis in Alagille syndrome: Results of a multicenter observational study. Hepatol Commun 2020;4:387-98.
- Kamath BM, Yin W, Miller H, Anand R, Rand EB, Alonso E, et al. Outcomes of liver transplantation for patients with Alagille syndrome: The studies of pediatric liver transplantation experience. Liver Transpl 2012;18:940-8.
- Arnon R, Annunziato R, Miloh T, Suchy F, Sakworawich A, Hiroshi S, et al. Orthotopic liver transplantation for children with Alagille syndrome. Pediatr Transplant 2010;14:622-8.
Tags: Miscellaneous